
About Fabry
Fabry Disease
Fabry disease is a lysosomal storage disorder named after the physicians who first described it in the late 19th century.
In 1898, two German doctors independently reported cases of the disease: Johann Fabry, a dermatologist, described patients with small, dark red or purple skin lesions (angiokeratomas). William Anderson, a British physician, reported similar symptoms, including kidney and nervous system involvement.
Because both described the same condition around the same time, it is sometimes called Anderson-Fabry disease.
What causes Fabry?
Fabry disease results from abnormal deposits of a particular fatty substance (called globotriaosylcera-mide) in blood vessel walls throughout the body. The primary defect which allows this to occur is the inherited deficiency of the enzyme, alpha galactosidase A, which is normally responsible for the breakdown of globotriaosylceramide.
Disease Inheritance
Fabry disease is an X-linked disorder and can be passed to children by either parent.
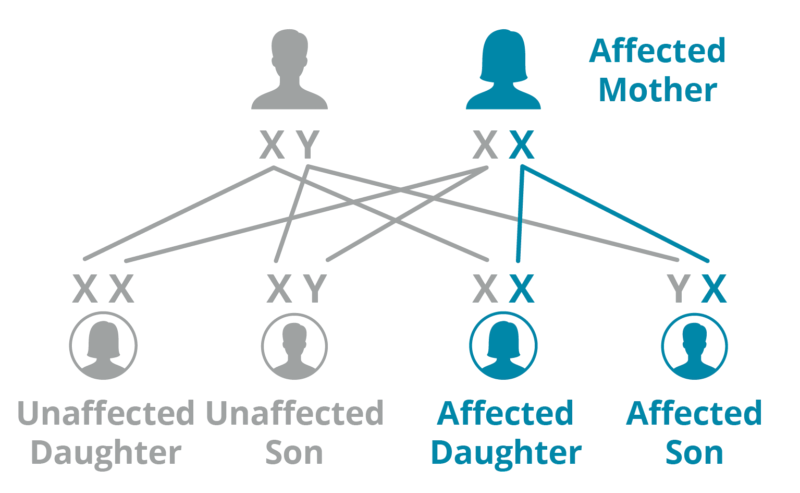
Mother
A mother with Fabry has a 50% chance of passing her X mutation to any of her children.
Father
A father with Fabry passes his X mutation to all of his daughters. His sons do not inherit Fabry because they inherit his Y chromosome.
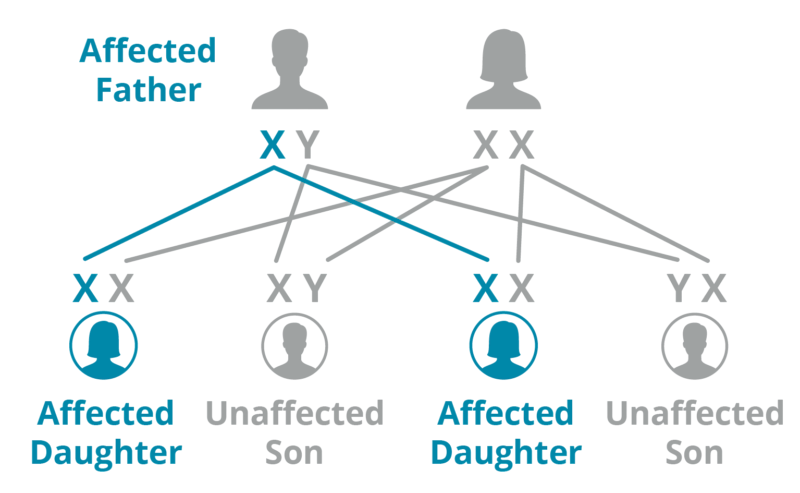
*Graphic used with permission by Fabry International Network.
It is estimated that one person in 40,000 has classic Fabry disease.
Clinical Symptoms
Fabry disease symptoms vary depending on the type. Some symptoms are mild and might not appear until later in life.

BRAIN & NERVES
- Burning in hands/feet
- Sensitivity to heat/cold
- Mini-stroke or stroke
- White matter lesions
- Dizziness
- Neuropathic pain
HEART
- Enlarged heart (LVH - Left ventricular hypertrophy)
- Heart failure
- Irregular heartbeat (Arrhythmia)
- Risk of heart attack

KIDNEYS
- Reduced kidney function
- Kidney failure (Decreased eGFR)
- Protein in urine (Proteinuria)
EYES & EARS
- Cloudy vision / cataracts
- Corneal whorls
- Hearing loss
- Ringing in ears (Tinnitus)
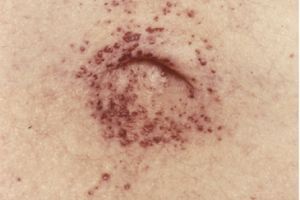
SKIN
- Red/purple spots (Angiokeratomas)
- Reduced or excessive sweating (Hypohydrosis or hyperhyrdrosis)
RESPIRATORY
- Shortness of breath
- Wheezing
- Fatigue

GASTROINTESTINAL
- Nausea / vomiting
- Diarrhea / constipation
- Abdominal cramps / bloating
- Food intolerance

PSYCHOLOGICAL
- Anxiety
- Depression
- Emotional distress
Treatments
There is no cure for Fabry disease but current treatments may prevent organ damage and greatly improve the quality of life of patients. Below is a list of treatments currently approved for use in the United States.
Enzyme Replacement Therapy (ERT)
Enzyme replacement therapy is a treatment that helps replace the missing or deficient enzyme (α-galactosidase A) in people with Fabry disease. Delivered through intravenous infusions, ERT works by breaking down the harmful substances that build up in the body’s cells, helping to protect the heart, kidneys, nervous system, and other organs. Treatments are usually given every two weeks, and while ERT does not cure Fabry disease, it can reduce symptoms, slow disease progression, and improve quality of life.
Fabrazyme® (Agalsidase Beta)
Company: Sanofi / Genzyme
How It Works:
Fabrazyme is an intravenous enzyme replacement therapy that provides the missing α-galactosidase A enzyme. It helps break down the substances that accumulate in cells, protecting the heart, kidneys, and nervous system.
Dosing: Typically administered
every two weeks.
Elfabrio® (Pegunigalsidase Alfa)
Company: Chiesi Global Rare Diseases / Protalix BioTherapeutics
How It Works:
Elfabrio is an intravenous, long-acting enzyme replacement therapy that stabilizes and prolongs enzyme activity in the bloodstream, helping to break down accumulated cellular substances.
Dosing: Typically administered
every two weeks.
Oral Chaperone Therapy
Some forms of Fabry disease stem from genetic changes that cause a key enzyme to misfold and stop working. Oral chaperone therapy helps the body fold and stabilize this enzyme so it can reach the right parts of the cell and break down harmful buildup. Taken by mouth, this personalized treatment works only for specific genetic variants and offers a convenient alternative to intravenous therapy.
Galafold® (Migalastat)
Company: Amicus Therapeutics
How It Works:
Galafold is an oral pharmacological chaperone therapy that helps stabilize certain mutant forms of the α‑galactosidase A (α‑Gal A) enzyme in people with Fabry disease, allowing their own enzyme to fold properly and break down harmful fatty substances in the cell.
Who It’s For: Adults with Fabry disease who have a confirmed amenable GLA gene variant.
Dosing: Take one 123 mg capsule
every other day, at the same time of day, on an empty stomach (fast 2 hours before and 2 hours after dosing).
Emerging Treatments
new oral medicines
Options that reduce harmful buildup or help stabilize the enzyme
LEARN MORE
Gene Therapy
Being studied as a possible one-time treatment to help the body make its own enzyme
mRna-based therapies
May encourage the body to produce more of the needed enzyme
LEARN MORE



